REGISTRATION – Version 4.12, released May 2025
______________________________________________
What’s new in Version 4.12
- Porting for LAMMPS simulation package (data and input files).
- Automatic single-point calculations in LAMMPS (if supplied) after conversion.
- New force field schemes – TraPPE united atom model.
- General force field libraries update.
- Include CMAP features (applicable to CHARMM22_prot and CHARMM36_prot), for GROMACS output.
________________________________________________
DL_FIELD is a utility software tool to setup force field (FF) models for molecular simulations such as the molecular dynamics (MD).
Software Development Strategy
To develop a user-friendly software tool that automatically process the complex molecular information with minimum user’s intervention and to speed up scientific outputs.
Primary Functions
- Force field model convertor: Single-step conversion of user’s molecular configuration into FF model files for DL_POLY, GROMACS and LAMMPS MD software packages based on user-selectable FF scheme.
- Force field editor: Edit or modify a standard FF scheme to produce customised schemes that is specific to a particular type of molecular model.
- Natural atom types descriptor: Automatic identification of chemical natural of every atom in the system.
- Available force fields: CHARMM, Amber, OPLS, TraPPE, PCFF, CVFF, COMPASS, Inorganic (oxides, clays, glass, zeolites), MISC_FF, etc.
Unique Capabilities
- Universal atom typing (DL_F Notation). Include chemical metadata directly into simulations for results analysis.
- Integration of FF schemes into identical DL_FIELD format.
- Multiple FF model setup such as bio-inorganic models.

Selected Example models
Applicable to a wide range of models – proteins, nuclei acids, carbohydrates, drug molecules, complex networked structure, polymers, inorganic solids, minerals and the mixture thereof.

Complex organic systems
C. W. Yong, ‘Study of interactions between polymer nanoparticles and cell membranes at atomistic levels’ Phil. Trans. R. Soc. B 370, 20140036 (2015)

General organic and biomolecules
O.J. Hills, C.W. Yong, A.J. Scott, D.A. Devine, J. Smith, H.F. Chappell, ‘Atomic-scale interactions between quorum sensing autoinducer molecules and the mucoid P. aeruginosa exopolysaccharide matrix’ Sci. Rep. 12, 7724 (2022).
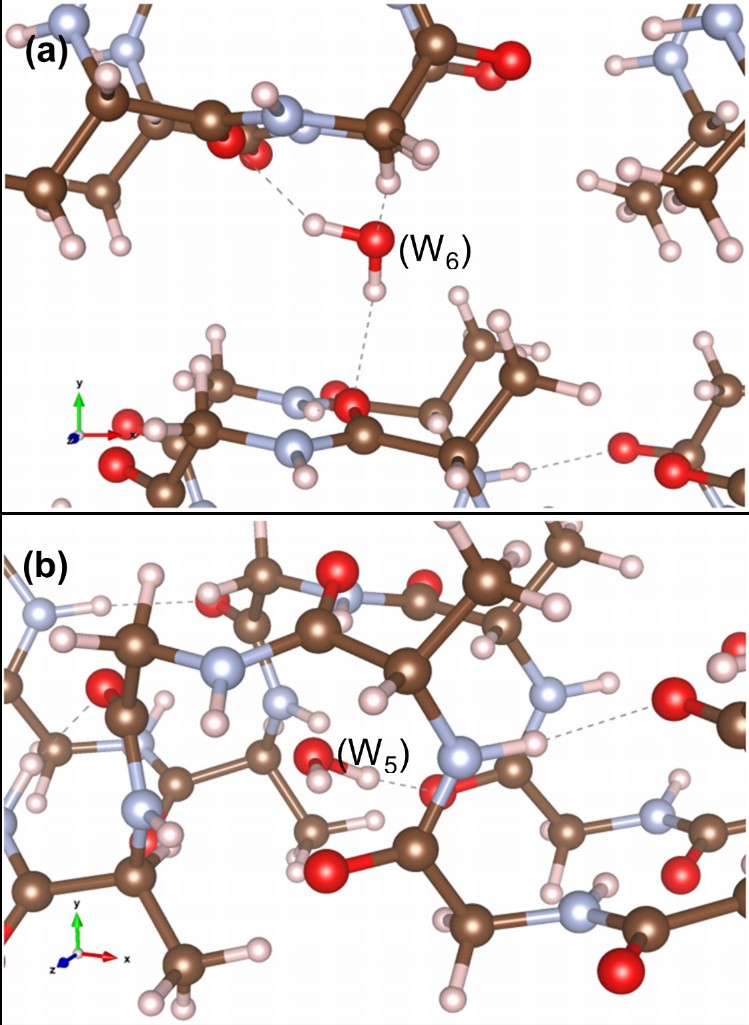
Protein-based systems
M.J. Haskew, B. Deacon, C.W. Yong, J.G. Hardy and S.T. Murphy, ‘Atomistic Simulation of water incorporation and mobility in bombyx mori silk fibrion’, ACS Omega 6, 35494-35504 (2021)

Nano-composite materials
L. Kong, R. Alqus, C.W. Yong, I.T. Todorov, S.J. Eichhorn, R.A. Bryce, ‘Cellulose I-beta microfibril interaction with pristine graphene in water: Effects of amphiphilicity by molecular simulations’, J. Mole. Graphics Model. 118, 108336 (2023)
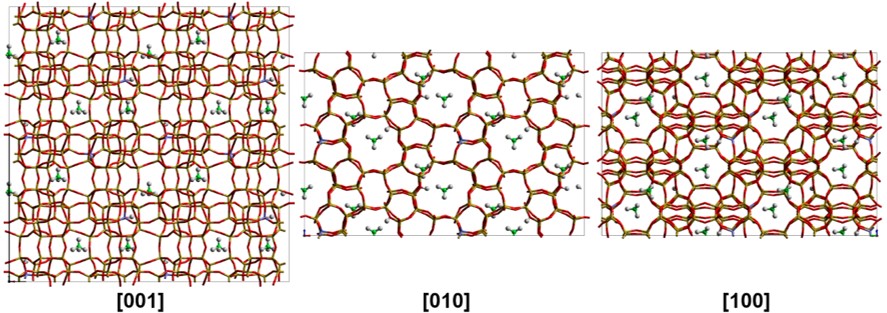
Porous materials
A.P. Hawkins, A. Zachariou, I.P. Silverwoord, et. al. ‘Combining quasielastic neutron scattering and molecular dynamics to study methane motions in ZSM-5’, J. Chem. Phys. 157, 184702 (2022)
Relevant Links
DL_POLY particle-based dyanamics simulation software
D_ATA – Atom Typer and Anayser: use of DL_F Notation to identify, quantify and annotate atomic interactions.
DL_FIELD User Manual, Ver 4.13 (June 2026)
DL_FIELD additional information, at DL_Software Digital Guide (DL_SDG)
DL_FIELD Resource Guide – Features illustrations. Available at DL_Software Digital Guide (DL_SDG)
DL_FIELD Tutorial Exercises, at DL_SDG.
DL_FIELD Tutorials for beginners – Step-by-step guide for simple polymeric systems setup. For DL_POLY, GROMACS and LAMMPS.
DL_F Notation:
- Journal reference: C.W. Yong, ‘Description and implementations of DL_F Notation: A natural chemical expression system of atom types for molecular simulations’, J. Chem. Inf. Model. 56, 1405-1409 (2016)
- Additional information, available at SSRN: http://dx.doi.org/10.2139/ssrn.4254942
DL_FIELD references
If you use DL_FIELD in your work, please quote the following reference:
C.W. Yong, ‘Description and implementations of DL_F Notation: A natural chemical expression system of atom types for molecular simulations’, J. Chem. Inf. Model. 56, 1405-1409 (2016)